 Home Visit
Home Visit Upload
Upload



Preventive Healthcare
Exploring Pheochromocytoma: Symptoms, Diagnosis, And Treatment
1172 Views
0
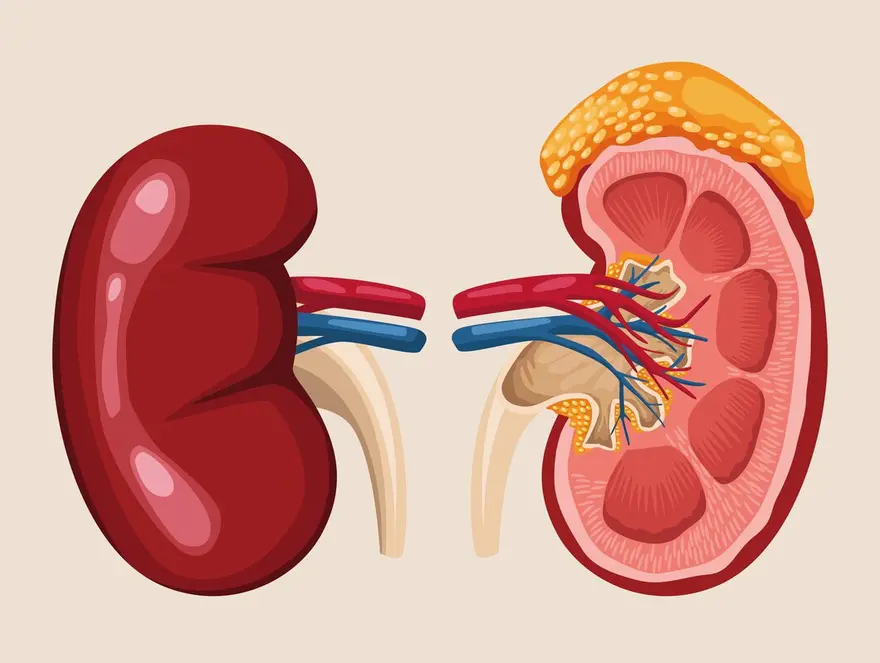
What Is Pheochromocytoma?
Pheochromocytoma is a rare type of tumour that arises from the chromaffin cells or neuroendocrine cells (hormone-producing glands) of the adrenal gland, which is located on top of each kidney. These tumours predominantly produce excessive amounts of catecholamines (neurotransmitters), which include adrenaline and noradrenaline. The secretion of these hormones can lead to various symptoms. Pheochromocytomas are generally benign (non-cancerous) but can be malignant (cancerous) in a small percentage of cases.
These tumours can occur at any age but are most commonly diagnosed in adults during their 30s to 50s. Pheochromocytomas may occur as part of hereditary syndromes such as multiple endocrine neoplasia type 2 (MEN2), von Hippel-Lindau disease, and neurofibromatosis type 1.
What Are The Symptoms Of Pheochromocytoma?
Key pheochromocytoma symptoms include:
- High blood pressure (hypertension).
- Headache.
- Excessive sweating for no known reason.
- A pounding, fast or irregular heartbeat.
- Feeling shaky
- Pain in your chest and/or abdomen.
- Being much paler than usual.
- Nausea and/or vomiting.
- Diarrhea.
- Constipation.
- An extreme drop in blood pressure upon standing suddenly (orthostatic hypotension).
- Unexplained weight loss
What Causes Pheochromocytoma?
The exact pheochromocytoma causes are not well understood, but several genetic factors contribute to its development:
- Multiple endocrine neoplasia 2 syndrome (a rare condition caused by a genetic mutation that affects multiple glands in your endocrine system), types A and B (MEN2A and MEN2B).
- Von Hippel-Lindau (VHL) disease (a rare genetic disorder that significantly increases the chance of having certain kinds of cancerous (malignant) tumours and noncancerous (benign) tumours and cysts).
- Neurofibromatosis type 1 (NF1) (a condition that affects your skin and nervous system (brain, spinal cord and nerves))
- Hereditary paraganglioma syndrome (a genetic condition that predisposes individuals to developing rare tumours called paragangliomas).
- Carney-Stratakis dyad [paraganglioma (rare tumours that arise from cells of the peripheral nervous system) and gastrointestinal stromal tumour (GIST) (tumours that develop in the walls of the stomach and intestines)].
How Is Pheochromocytoma Diagnosed?
Pheochromocytoma diagnosis involves a combination of clinical assessment, biochemical tests, and imaging studies to confirm the presence of the tumour and its activity. The pheochromocytoma diagnosis typically includes:
- Clinical Evaluation: Review of symptoms and medical history.
- Biochemical Testing: Measurement of catecholamines and metanephrines in blood and urine.
- Imaging Studies:
- CT Scan: Often the first imaging technique used.
- MRI: Used for its superior soft tissue contrast.
- MIBG Scan: Detects extra-adrenal (outside adrenal glands) and metastatic (cancerous) tumours.
- Genetic Testing: Recommended for suspected hereditary cases.
What Are The Risk Factors Of Pheochromocytoma?
The risk factors for developing pheochromocytoma include:
- Genetic Disorders: Having a hereditary syndrome such as Multiple Endocrine Neoplasia type 2 (MEN2), von Hippel-Lindau disease, neurofibromatosis type 1, or hereditary paraganglioma-pheochromocytoma syndromes significantly increases the risk.
- Family History: A family history of pheochromocytoma or related genetic conditions elevates the likelihood of developing the tumour.
- Genetic Mutations: Specific mutations in genes like RET, VHL, NF1, and SDHx are linked to a higher risk of pheochromocytoma.
What Are The Complications Of Pheochromocytoma?
Pheochromocytoma causes several serious complications if not diagnosed and treated promptly:
- Severe Hypertension: Persistently high blood pressure or sudden severe spikes can lead to cardiovascular complications such as heart attack, stroke, or heart failure.
- Cardiac Arrhythmias: Abnormal heart rhythms, which can be life-threatening, may be triggered by excessive catecholamine levels.
- Heart Damage: Chronic high levels of catecholamines can lead to cardiomyopathy (disease of the heart muscle), impairing the heart's ability to pump blood effectively.
- Crisis with Multiorgan Failure: A pheochromocytoma crisis, often triggered by stress, surgery, or certain medications, can cause severe hypertension, leading to multiorgan failure or even death if not treated immediately.
- Metastatic Disease: Although rare, pheochromocytomas can metastasise (spread to other parts of the body), complicating pheochromocytoma treatment and worsening prognosis.
- Adverse Effects from Surgery: Surgical removal of the tumour, while often curative, carries risks of complications, especially if the tumour is not diagnosed preoperatively and managed appropriately.
What Tests Are Used To Diagnose Pheochromocytoma?
Diagnosing pheochromocytoma typically involves a combination of biochemical testing and imaging techniques to detect and locate the tumour. Commonly used tests are:
Biochemical Tests
- Plasma-Free Metanephrines: This test measures the levels of metanephrines (metabolites of catecholamines) in the blood. It is highly sensitive and can detect tumours that secrete catecholamines episodically.
- 24-Hour Urinary Catecholamines and Metanephrines: This involves collecting urine for 24 hours to measure the levels of catecholamines and their metabolites. It helps confirm the production of excess hormones typical of pheochromocytomas.
Pheochromocytomas Radiology (Imaging) Studies
- Computed Tomography (CT) Scan: A CT scan of the abdomen is typically the first imaging test performed to locate an adrenal tumour.
- Magnetic Resonance Imaging (MRI): MRI is used for patients where radiation exposure is a concern or for better soft tissue contrast. It’s particularly useful in detecting small or extra-adrenal pheochromocytomas.
- Metaiodobenzylguanidine (MIBG) Scintigraphy: This nuclear medicine scan is used if CT or MRI results are inconclusive.
Additional Diagnostic Tests
- Genetic Testing: Given the association with genetic syndromes, testing for mutations in genes such as RET, VHL, NF1, and SDHx is recommended for patients with a family history of pheochromocytoma or related conditions.
- Clonidine Suppression Test: Rarely used, but can be helpful in ambiguous cases. It measures plasma catecholamines before and after administration of clonidine, which should normally suppress norepinephrine in non-pheochromocytoma cases.
How Is Pheochromocytoma Treated?
Pheochromocytoma treatment varies depending on the specifics of the case, such as whether the tumour is benign or malignant, and if it has metastasised. Here are the main pheochromocytoma treatment options available:
1. Surgery
- Primary Treatment: Surgery is the primary pheochromocytoma treatment and is often curative, especially when the tumour is localised to the adrenal glands.
- Laparoscopic Adrenalectomy: This minimally invasive procedure is commonly used to remove single, small, benign tumours.
- Open Adrenalectomy: Required for larger or invasive tumors, or when malignancy is involved.
2. Radiation Therapy
- Palliative Care: Used mainly for pain relief in cases of bone metastasis from malignant pheochromocytoma.
3. Chemotherapy
- For Malignant Cases: Used if the tumour is malignant or has metastasised and is not amenable to complete surgical resection. Common chemotherapy agents include cyclophosphamide, vincristine, and dacarbazine.
4. Ablation Therapy
- Radiofrequency Ablation (RFA): Used for small or inoperable tumours to destroy cancer cells with heat.
5. Embolization Therapy
- Pre-Surgical or Palliative: This procedure involves blocking the blood supply to the tumour, often used preoperatively to shrink the tumour or palliatively to manage pheochromocytoma symptoms.
6. Targeted Therapy
- MIBG Therapy: Uses a radioactive iodine-labeled form of MIBG that targets catecholamine-producing cells, useful particularly in cases where traditional chemotherapy is less effective.
- Targeted Molecular Therapies: These are newer treatments that target specific pathways or mutations in tumour cells and are considered when other treatments fail or aren’t suitable.
What Is The Outlook For Pheochromocytoma?
The outlook for pheochromocytoma is generally positive when it receives proper treatment. Surgery successfully removes about 90% of these tumours.
However, if pheochromocytomas are not treated, they can lead to severe, potentially fatal complications, including:
- Cardiomyopathy (heart muscle disease).
- Myocarditis (inflammation of the heart muscle).
- Cerebral haemorrhaging (uncontrolled bleeding in the brain).
- Pulmonary edema (fluid accumulation in the lungs).
Additionally, untreated pheochromocytomas can increase the risk of experiencing a stroke or heart attack (myocardial infarction).
Conclusion
In conclusion, while pheochromocytoma can pose serious health risks if left untreated, the prognosis is typically very good when diagnosed early and managed effectively with surgical removal. Around 90% of these tumours can be successfully excised, significantly reducing the risk of complications of pheochromocytoma. However, it is crucial to recognise and address the potentially life-threatening complications associated with untreated pheochromocytomas, such as heart disease, stroke, and severe cardiovascular events.
Metropolis Healthcare provides a comprehensive range of 4000+ clinical laboratory tests and profiles. Schedule your appointment with Metropolis Healthcare today and experience comprehensive care and expert diagnostics. Let's get started!







1701259759.webp)









 WhatsApp
WhatsApp